
近年來 RNA-seq 已成為分子生物學中不可或缺的工具,對於各物種的轉錄體 ( Transcriptome ) 提供了更深入的理解。然而,在過去十多年的發展中,儘管在揭示基因表現方面取得巨大的進展,卻始終無法準確解答轉錄本 ( Transcript ) 的真正結構,以及不同分析工具估算出的基因或是轉錄本的表現量。PacBio 公司的 RNA 全長定序 ( Iso-Seq ),這項技術使我們能夠直接獲得長達 10kb 以上的轉錄異構體 ( isoforms ) 的外顯子結構 ( exonic structure ),而無須經過後續分析工具的組裝,並有助於準確地量化基因與轉錄本的表現量,使我們能夠更精確地計算差異表現基因 ( Differential Expression Gene, DEG )、差異表現轉錄本 ( Differential Expression Transcript, DET ) 以及差異轉錄本使用 ( Differential Transcript Usage, DTU ) 等分析。
Iso-Seq 的優勢總結如下:
基因表現分析結果的平台差異:短片段 RNA-seq vs. 長片段分析 Iso-Seq
阿德雷德 ( Adelaide ) 大學與 PacBio[1] 於 2023 年發表的研究中,專注於揭示安格斯牛 ( Angus ) 與婆羅門牛 ( brahman ) 中複雜的轉錄差異。因此以胎兒發育中期 ( 約153天 ) 的肝臟 ( liver )。研究利用 RNA-seq 與 Iso-Seq 平台的定序,分別進行三組重複樣品的定序。為了提升 Iso-Seq 平台的數據量,樣品都使用了兩個 SMRT cells 進行定序。研究採用安格斯牛 x 婆羅門牛雜交第一代 ( GCF_003369695.1 ) 作為參考基因組進行分析,Iso-Seq 分析結果中發現了 54,692 個新的 isoform。在基因的部分,無論 RNA-seq 或是 Iso-Seq 的樣品在相同平台的表現量都呈現高度正相關性,兩個亞種之間的相關係數都能夠有 0.9 以上 ( 圖二 )。然而,若使用不同定序平台,基因表現量的相關係數會降至 0.7 至 0.8 ( 圖二 )。值得注意的是,在轉錄本的水平上,相關係數更是驟降至 0.2 至 0.3 ( 圖三 )。作為 Iso-Seq 主要研究標的 isoforms 上,可以確定 Iso-Seq 與 RNA-seq 的分析結果在轉錄體上存在明顯的不同,就算在基因層面,相似程度也有所不同了。接著探討兩個平台表現量差異對於 DEG 與 DET 產生的影響。
根據 RNA-seq 的相關研究,差異表現工具對於結果的影響高於序列比對工具[2]。在表現量有明顯差異的情況下,Iso-Seq DEG 與 RNA-seq DEG ( count ) 都使用同樣檢定工具-limma,但兩者共同顯著的基因也僅有 37 個。因此,需要近一步檢視兩個平台數據差異的原因為何。
基因表現分析結果平台差異的原因來源
另一個問題-歸屬基因無法確定,是指一基因組區間中有多個基因重疊於此。這問題作者使用一個基因本身在其他基因上重疊的程度來評估,結果顯示,Iso-Seq 中顯著的基因有較多與其他基因重疊的比例較高 ( 圖五 ),然而根據 RNA-seq 分析軟體 FeatureCounts 默認的條件,當 reads 被定位在這些不明的基因重疊區域時會被忽略不計算,表示這些基因在短片段的 RNA-seq 研究中,容易受到歸屬基因不明的影響,而增加偽陰性的風險。
由於前述的分析顯示 Iso-Seq 不受多重定位及歸屬基因無法確定的影響,因此能夠準確呈現 isoforms 的表現量,進而準確的分析樣品在不同條件或狀態下的轉錄異構體切換 ( isoform switch )。這方面的分析通常稱為差異轉錄本使用 ( Differential transcript usage, DTU ) 分析,其功能主要檢定同一基因內不同 isoforms 的組成變化。根據 Iso-Seq 進行的 DTU 分析的結果,其中 MRPL49 在 Iso-Seq 的 DTU 分析中呈現顯著差異 ( 圖六 ),表示 MRPL49 中的 isoforms 在不同亞種的比例有顯著的不同。其中 PB.27455.10 主要發生在安格斯牛;而 PB.27455.1 則反之。通過 PB.27455.10 及 PB.27455.1 的外顯子結構表示兩者之間僅有在 5' 端存在 52bp 的差異,而這樣的差異在 RNA-seq 的資料中是不容易被發現。
儘管 Iso-Seq 解決了 RNA-seq 普遍面臨的問題,也驗證了單獨 RNA-seq 的分析可能導致結果的偏誤。作者同時提了其他可能導致兩者差異的原因。首先,Iso-Seq 在進行擴增時,主要的擴增範圍可達數百甚至數千個鹼基 ( base )。然後在建立文庫的過程中,根據使用的 kit 的不同,會對 RNA 進行不同大小的篩選。最後,由於 SMRT cells 上 ZMWs 孔洞的限制,導致豐富度較低的 RNA 在上機過程中被濾去。儘管如此,Iso-Seq 還是較能呈現基因在轉錄過程中的複雜性。若長片段定序的能夠增加數據量,將能夠更深入了解 RNA 特性和基因表達的例行定量分析的準確性。
長片段定序增加數據量的解方 - 以單細胞轉錄組分析為例
在得到每個 cDNA 串接的序列後,首先需要將所得的長讀長拆分成各自的初始 cDNA 片段,而拆分主要是藉由辨識以下序列特徵 ( 圖八 ):
MAS-ISO-seq 於鑑別基因表現差異的實例驗證

【參考資料】
1. Tseng et al., (2023). Long read isoform sequencing reveals hidden transcriptional complexity between cattle subspecies. BMC genomics, 24(1), 1-15.
2. Munster et al., (2023). RNA-Seq Alignment and Differential Expression Software Comparison (No. DOT/FAA/AM-23/02). United States. Department of Transportation. Federal Aviation Administration. Office of Aviation. Civil Aerospace Medical Institute.
3. Robert, & Watson (2015). Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome biology, 16, 1-16.
4. Deschamps-Francoeur et al., (2020). Handling multi-mapped reads in RNA-seq. Computational and structural biotechnology journal, 18, 1569-1576.
5. Consiglio et al., (2016). A fuzzy method for RNA-Seq differential expression analysis in presence of multireads. BMC bioinformatics, 17, 95-110.
6. Al'Khafaji et al., (2023). High-throughput RNA isoform sequencing using programmed cDNA concatenation. Nat Biotechnol. Jun 8
7. 單細胞基因表現分析 Single cell gene expression profiling
8.【PacBio 第三代定序服務】Iso-Seq
9.【PacBio 第三代定序服務】16S full length rDNA Sequencing
Iso-Seq 的優勢總結如下:
- 能完整定序 mRNA isoform,且無須依賴組裝過程 ( 圖一 )。
- 每條序列能夠正確歸類於其所屬的 isoform,有助於分析轉錄體內的基因或是 isoforms 的表現特性。
- 即使在已知的物種中,仍能發現許多的未知的 genes 或是 isoforms。
- Iso-Seq 藉由讀長上的優勢,在沒有參考基因組 ( Reference genome ) 的狀況下,也有足夠的資訊進行功能預測。
- 在有 Reference genome 的樣品中,能夠獲得完整選擇性剪接 ( Alternative splicing ) 以及 基因融合 ( Gene Fusion ) 等信息。
 |
| 【圖一】RNA-seq 和 Iso-Seq 的讀長差異使得 Iso-Seq 可以發現較多數量的 isoform。 |
基因表現分析結果的平台差異:短片段 RNA-seq vs. 長片段分析 Iso-Seq
阿德雷德 ( Adelaide ) 大學與 PacBio[1] 於 2023 年發表的研究中,專注於揭示安格斯牛 ( Angus ) 與婆羅門牛 ( brahman ) 中複雜的轉錄差異。因此以胎兒發育中期 ( 約153天 ) 的肝臟 ( liver )。研究利用 RNA-seq 與 Iso-Seq 平台的定序,分別進行三組重複樣品的定序。為了提升 Iso-Seq 平台的數據量,樣品都使用了兩個 SMRT cells 進行定序。研究採用安格斯牛 x 婆羅門牛雜交第一代 ( GCF_003369695.1 ) 作為參考基因組進行分析,Iso-Seq 分析結果中發現了 54,692 個新的 isoform。在基因的部分,無論 RNA-seq 或是 Iso-Seq 的樣品在相同平台的表現量都呈現高度正相關性,兩個亞種之間的相關係數都能夠有 0.9 以上 ( 圖二 )。然而,若使用不同定序平台,基因表現量的相關係數會降至 0.7 至 0.8 ( 圖二 )。值得注意的是,在轉錄本的水平上,相關係數更是驟降至 0.2 至 0.3 ( 圖三 )。作為 Iso-Seq 主要研究標的 isoforms 上,可以確定 Iso-Seq 與 RNA-seq 的分析結果在轉錄體上存在明顯的不同,就算在基因層面,相似程度也有所不同了。接著探討兩個平台表現量差異對於 DEG 與 DET 產生的影響。
 |
| 【圖二】RNA-seq 及 Iso-Seq 在基因的層級分析,樣品在相同平台的表現量都呈現高度正相關性,兩個亞種之間的相關係數都能夠有 0.9 以上,若使用不同定序平台,基因表現量的相關係數降至 0.7 至 0.8 ( 取自 Tseng et al., 2023 )。 |
 |
| 【圖三】RNA-seq 及 Iso-Seq 在轉錄本的層級分析,樣品在相同平台的表現量都呈現高度正相關性,若使用不同定序平台,基因表現量的相關係數降至 0.2 至 0.3 ( 取自 Tseng et al., 2023 )。 |
根據 RNA-seq 的相關研究,差異表現工具對於結果的影響高於序列比對工具[2]。在表現量有明顯差異的情況下,Iso-Seq DEG 與 RNA-seq DEG ( count ) 都使用同樣檢定工具-limma,但兩者共同顯著的基因也僅有 37 個。因此,需要近一步檢視兩個平台數據差異的原因為何。
基因表現分析結果平台差異的原因來源
在量化 RNA-seq 數據時,常面臨缺乏足夠的訊息使 Reads 歸屬於一個基因的問題,尤其發生在多重定位 ( Multimapping ) 和歸屬基因無法確定的情況[3]。儘管 RNA-seq 的各種分析工具提供了不同策略,但這兩個問題仍然沒有得到解決[4]。相較之下,Iso-Seq 每條 Reads 都代表一個 isoform,因此無需進行組裝即可確定其所屬的基因。
首先多重定位是指一個序列重複出現在基因組的不同位置。從 Iso-Seq 的結果觀察到顯著的基因受到多重定位影響較小 ( 圖四 )。回顧 Consiglio 的研究[5]指出多重定位可能導致表現量高估,進而產生偽陽性的結果。而在 RNA-seq 結果為顯著的基因,受到多重定位影響的程度較高,這也可能增加了偽陽性的風險。
首先多重定位是指一個序列重複出現在基因組的不同位置。從 Iso-Seq 的結果觀察到顯著的基因受到多重定位影響較小 ( 圖四 )。回顧 Consiglio 的研究[5]指出多重定位可能導致表現量高估,進而產生偽陽性的結果。而在 RNA-seq 結果為顯著的基因,受到多重定位影響的程度較高,這也可能增加了偽陽性的風險。
 |
| 【圖四】以 Iso-seq 分析表現顯著差異的基因受到多重定位影響較小 ( 左及中 ),而短片段 RNA-seq 受到多重定位影響較大 ( 取自 Tseng et al., 2023 )。 |
另一個問題-歸屬基因無法確定,是指一基因組區間中有多個基因重疊於此。這問題作者使用一個基因本身在其他基因上重疊的程度來評估,結果顯示,Iso-Seq 中顯著的基因有較多與其他基因重疊的比例較高 ( 圖五 ),然而根據 RNA-seq 分析軟體 FeatureCounts 默認的條件,當 reads 被定位在這些不明的基因重疊區域時會被忽略不計算,表示這些基因在短片段的 RNA-seq 研究中,容易受到歸屬基因不明的影響,而增加偽陰性的風險。
 |
| 【圖五】以基因彼此重疊程度來評估 RNA-seq 及 Iso-Seq 發現的表現顯著差異基因具有歸屬基因不明的數量。結果顯示,Iso-Seq 中顯著的基因較多和其他基因重疊的比例較高,推測是因 Iso-Seq 技術較不受歸屬基因不明的影響 ( 取自 Tseng et al., 2023 )。 |
由於前述的分析顯示 Iso-Seq 不受多重定位及歸屬基因無法確定的影響,因此能夠準確呈現 isoforms 的表現量,進而準確的分析樣品在不同條件或狀態下的轉錄異構體切換 ( isoform switch )。這方面的分析通常稱為差異轉錄本使用 ( Differential transcript usage, DTU ) 分析,其功能主要檢定同一基因內不同 isoforms 的組成變化。根據 Iso-Seq 進行的 DTU 分析的結果,其中 MRPL49 在 Iso-Seq 的 DTU 分析中呈現顯著差異 ( 圖六 ),表示 MRPL49 中的 isoforms 在不同亞種的比例有顯著的不同。其中 PB.27455.10 主要發生在安格斯牛;而 PB.27455.1 則反之。通過 PB.27455.10 及 PB.27455.1 的外顯子結構表示兩者之間僅有在 5' 端存在 52bp 的差異,而這樣的差異在 RNA-seq 的資料中是不容易被發現。
 |
| 【圖六】基因 MRPL49 在不同樣品的轉錄本之外顯子結構差異 ( 取自 Tseng et al., 2023 )。 |
儘管 Iso-Seq 解決了 RNA-seq 普遍面臨的問題,也驗證了單獨 RNA-seq 的分析可能導致結果的偏誤。作者同時提了其他可能導致兩者差異的原因。首先,Iso-Seq 在進行擴增時,主要的擴增範圍可達數百甚至數千個鹼基 ( base )。然後在建立文庫的過程中,根據使用的 kit 的不同,會對 RNA 進行不同大小的篩選。最後,由於 SMRT cells 上 ZMWs 孔洞的限制,導致豐富度較低的 RNA 在上機過程中被濾去。儘管如此,Iso-Seq 還是較能呈現基因在轉錄過程中的複雜性。若長片段定序的能夠增加數據量,將能夠更深入了解 RNA 特性和基因表達的例行定量分析的準確性。
長片段定序增加數據量的解方 - 以單細胞轉錄組分析為例
RNA 基因表現的動態範圍 ( dynamic range ) 為-107-108,一般短片段 RNA-seq 定序量多為 20 M reads,動態範圍約為 107;而 PacBio flowcell 的產能輸出相對較低,約為 2M ~ 6M reads,限制了該平台在基因表現生物問題的適用性。
因此,許多方法學研發用以解決如何提升每個奈米孔每次可讀取的序列分子數,其中於 2023 年發表的 Multiplexed ArrayS isoform sequencing ( 以下簡稱 MAS-ISO-seq ) 方法可以同時解析 RNA 轉錄異構體全長,並且產量提高 15 倍[6]!
使用分子間串接組合以提高產量
因此,許多方法學研發用以解決如何提升每個奈米孔每次可讀取的序列分子數,其中於 2023 年發表的 Multiplexed ArrayS isoform sequencing ( 以下簡稱 MAS-ISO-seq ) 方法可以同時解析 RNA 轉錄異構體全長,並且產量提高 15 倍[6]!
使用分子間串接組合以提高產量
這種方法的核心設計是將多個 cDNA 序列串接組合成一個分子 ( 稱之為「陣列」(array) ),再進行定序,然後將所得的序列拆解還原成其原來 cDNA 片段以進行分析。
於建構好 cDNA 文庫後,將其平行分成數個 PCR 反應。在每個 PCR 反應中,加入不同的條碼序列以連接在 cDNA 的兩端,其中設計的巧妙之處在於,條碼序列的其中一股的組成核苷酸帶有 deoxy-uridine(dU),經由酵素將帶有 dU 的該股水解後,使得末端並不是平的、而是「突出」的,且這些「突出」的 DNA 序列在平行的 PCR 反應中為互補序列,因此在將兩端連接好不同的條碼序列 cDNA 匯集在一個反應管時,這些互補的兩端會將 cDNA 分子頭尾相「黏」而串接成一更長的分子 ( 圖七 ),陣列中的條碼是已知的 DNA 連接反應順序 ( A、B、C、D、E 等 ),由於定序長度上限為 25000bp、若平均 cDNA 分子長度為 1500bp,則可推算出並行 PCR 反應數量的上限約為 16 個反應。
於建構好 cDNA 文庫後,將其平行分成數個 PCR 反應。在每個 PCR 反應中,加入不同的條碼序列以連接在 cDNA 的兩端,其中設計的巧妙之處在於,條碼序列的其中一股的組成核苷酸帶有 deoxy-uridine(dU),經由酵素將帶有 dU 的該股水解後,使得末端並不是平的、而是「突出」的,且這些「突出」的 DNA 序列在平行的 PCR 反應中為互補序列,因此在將兩端連接好不同的條碼序列 cDNA 匯集在一個反應管時,這些互補的兩端會將 cDNA 分子頭尾相「黏」而串接成一更長的分子 ( 圖七 ),陣列中的條碼是已知的 DNA 連接反應順序 ( A、B、C、D、E 等 ),由於定序長度上限為 25000bp、若平均 cDNA 分子長度為 1500bp,則可推算出並行 PCR 反應數量的上限約為 16 個反應。
 |
| 【圖七】MAS-ISO-seq 的實驗流程 ( 取自 Al'Khafaji et al., 2023 )。 |
串接序列的拆分
- cDNA 分子序列特徵,如:polyA tail、TSO 序列。
- cDNA 文庫製備時的 adaptor 序列,且陣列中的條碼是已知的 DNA 連接反應順序 ( A、B、C、D、E 等 )。
 |
| 【圖八】MAS-ISO-seq 序列的結構。 |
MAS-ISO-seq 於鑑別基因表現差異的實例驗證
如此,MAS-ISO-seq 既可以分析全長轉錄組用可以利用其增加的數據進行基因表現差異分析。以 6,000 個腫瘤浸潤細胞毒性 T 細胞的單細胞轉錄分析,該文庫既使用短讀長定序,又進行 MAS-ISO-seq 實驗。圖九A - 為短讀長定序的結果,其中細胞清楚地分成代表休眠/活化/耗盡狀態的分群狀態。
將 MAS-ISO-seq 結果從 33M reads 向下採樣到 1.6M reads 時,可以看到數據量如何影響的群組分析的結果。隨著採樣數據的增加,細胞聚類變得更加穩定,越來越趨近於短片段的聚類 ( 圖九B、上及中圖 );且數據量越高時,可以找到顯著表現差異的 RNA 轉錄異構體的數量就越多 ( 圖九B、下圖 )。
將 MAS-ISO-seq 結果從 33M reads 向下採樣到 1.6M reads 時,可以看到數據量如何影響的群組分析的結果。隨著採樣數據的增加,細胞聚類變得更加穩定,越來越趨近於短片段的聚類 ( 圖九B、上及中圖 );且數據量越高時,可以找到顯著表現差異的 RNA 轉錄異構體的數量就越多 ( 圖九B、下圖 )。
 |
| 【圖九】短片段讀序及長片段讀序經電腦模擬下行採樣的群組分析結果比較。(A)以短片段定序、6,260 個 CD8+ T 細胞的單細胞 UMAP 基因表現群組分析結果,(B) 電腦模擬下行採樣的 MAS-ISO-seq 分析結果;( 上圖 ) 以 UMAP 呈現電腦模擬數據量多寡對群組分析結果的影響;( 中圖 ) 以 adjusted Rand index 分析短片段及 MAS-ISO-seq 結果,顯示 MAS-ISO-seq 的 read 數量越多,兩者的基因表現相似度越高;( 下圖 ) 數據量越高時,可以找到顯著表現差異的 RNA 轉錄異構體的數量就越多。( 取自 Al'Khafaji et al., 2023 )。 |
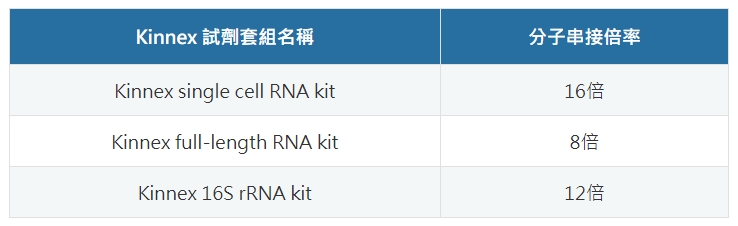
*PacBio 已於 2023 年底推出三款 Kinnex 試劑套組,大幅提高定序的通量
 |
| 【表一】PacBio MAS-ISO-seq 技術,分別針對單細胞 RNA、全長 RNA 和 16S rRNA 定序的通量,推出三種應用的試劑套組 - Kinnex;其中單細胞 RNA 串接倍數為 16 倍、全長 RNA 8 倍、16S rRNA 定序則為 12 倍。 |
【參考資料】
1. Tseng et al., (2023). Long read isoform sequencing reveals hidden transcriptional complexity between cattle subspecies. BMC genomics, 24(1), 1-15.
2. Munster et al., (2023). RNA-Seq Alignment and Differential Expression Software Comparison (No. DOT/FAA/AM-23/02). United States. Department of Transportation. Federal Aviation Administration. Office of Aviation. Civil Aerospace Medical Institute.
3. Robert, & Watson (2015). Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome biology, 16, 1-16.
4. Deschamps-Francoeur et al., (2020). Handling multi-mapped reads in RNA-seq. Computational and structural biotechnology journal, 18, 1569-1576.
5. Consiglio et al., (2016). A fuzzy method for RNA-Seq differential expression analysis in presence of multireads. BMC bioinformatics, 17, 95-110.
6. Al'Khafaji et al., (2023). High-throughput RNA isoform sequencing using programmed cDNA concatenation. Nat Biotechnol. Jun 8
7. 單細胞基因表現分析 Single cell gene expression profiling
8.【PacBio 第三代定序服務】Iso-Seq
9.【PacBio 第三代定序服務】16S full length rDNA Sequencing
留言
張貼留言